The goal of ibdfindr is to detect genomic regions shared identical by descent (IBD) between two individuals, using SNP genotypes. It does so by fitting a continuous-time hidden Markov model (HMM) to the data.
Until ibdfindr becomes available on CRAN, you may install the development version from GitHub:
remotes::install_github("magnusdv/ibdfindr")library(ibdfindr)As an example we consider the built-in dataset
cousinsDemo, which contains almost 4000 SNP genotypes for
two related individuals (in fact, a pair of cousins).
head(cousinsDemo)#> CHROM MARKER MB CM A1 A2 FREQ1 ID1 ID2
#> 1 1 rs9442372 1.083324 0.0000000 A G 0.3890 A/G G/G
#> 2 1 rs4648727 1.844830 0.1803812 A C 0.3628 A/C A/C
#> 3 1 rs10910082 2.487496 1.2670505 C T 0.3740 C/T C/T
#> 4 1 rs6695131 3.084360 1.9782286 C T 0.5719 C/T C/T
#> 5 1 rs3765703 3.675872 4.8403127 G T 0.4058 G/G T/T
#> 6 1 rs7367066 3.887191 5.7748481 C T 0.6739 C/C C/CThe function findIBD() conveniently wraps the key steps
of the package:
fitHMM())findSegments())ibdPosteriors())ibd = findIBD(cousinsDemo)
#> Individuals: ID1, ID2
#> Chromosome type: autosomal
#> Fitting HMM parameters...
#> Optimising `k1` and `a` jointly; method: L-BFGS-B
#> k1 = 0.185, a = 7.430
#> Finding IBD segments...
#> 14 segments (total length: 597.21 cM)
#> Calculating IBD posteriors...
#> Analysis complete in 0.937 secsFor details of the different steps, see the documentation of the
individual functions: fitHMM(),
findSegments(), and ibdPosteriors().
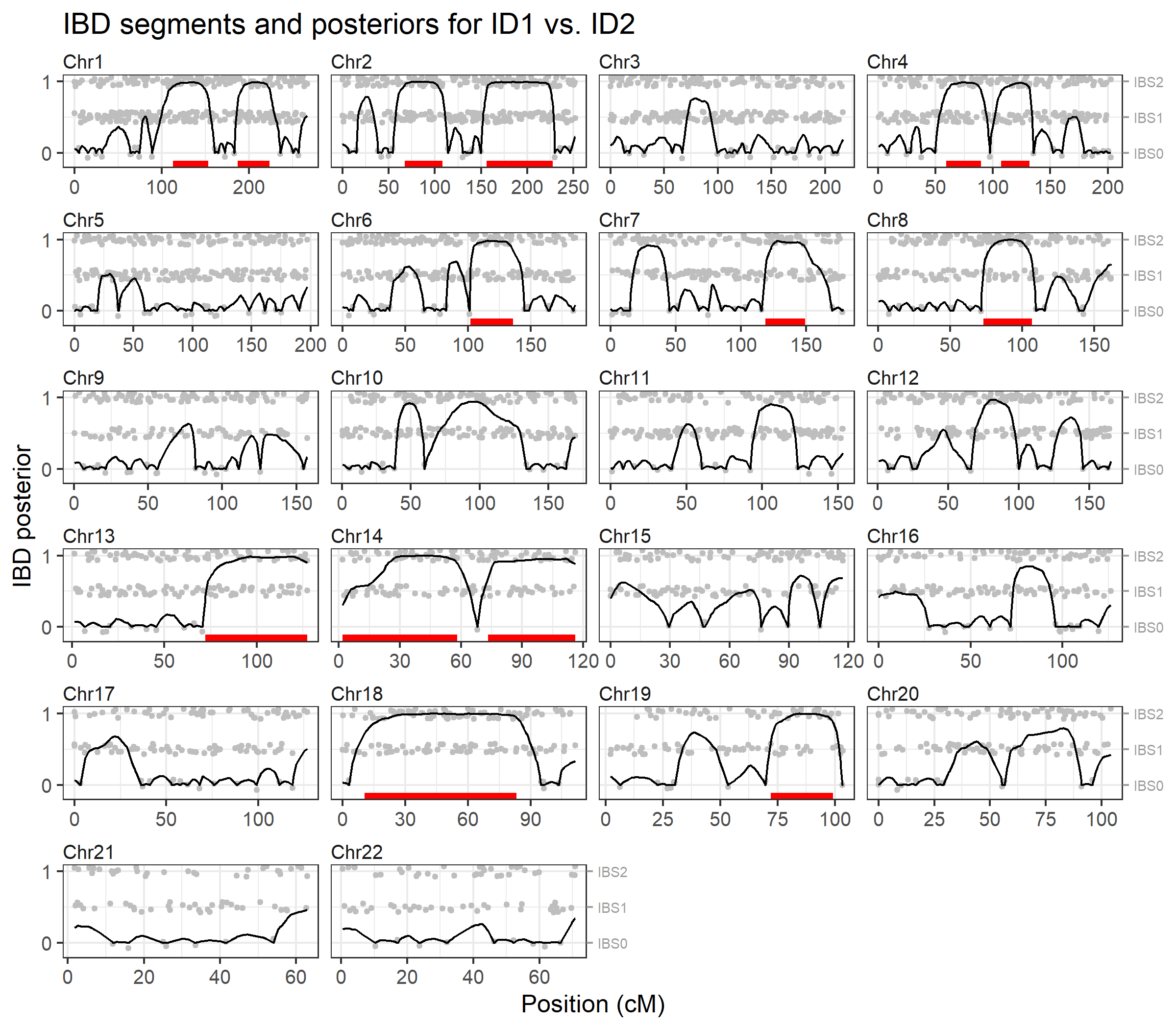
To visualise the results we pass the output to
plotIBD(). This plots the posterior probabilities on a
background (grey points) showing the identity-by-state (IBS) status at
each marker, i.e. whether the individuals have 0, 1 or 2 alleles in
common. Inferred IBD regions are shown as red segments at the bottom of
each chromosome panel.
plotIBD(ibd)
We may also inspect the identified segments as a data frame, showing the start and end positions, and the number of markers in each segment:
ibd$segments
#> chrom startCM endCM n
#> 1 1 113.000492 153.08623 54
#> 2 1 186.630319 223.39542 46
#> 3 2 67.118524 108.35905 50
#> 4 2 156.120361 227.62362 90
#> 5 4 59.474215 89.85522 41
#> 6 4 106.931772 131.69280 35
#> 7 6 102.041153 136.17734 43
#> 8 7 118.702115 149.11283 38
#> 9 8 72.976430 106.82971 55
#> 10 13 72.049460 127.22724 63
#> 11 14 1.808552 58.07117 67
#> 12 14 73.201009 116.00254 49
#> 13 18 10.592411 83.28443 101
#> 14 19 72.014020 99.15558 34The brothersX dataset contains genotypes for two
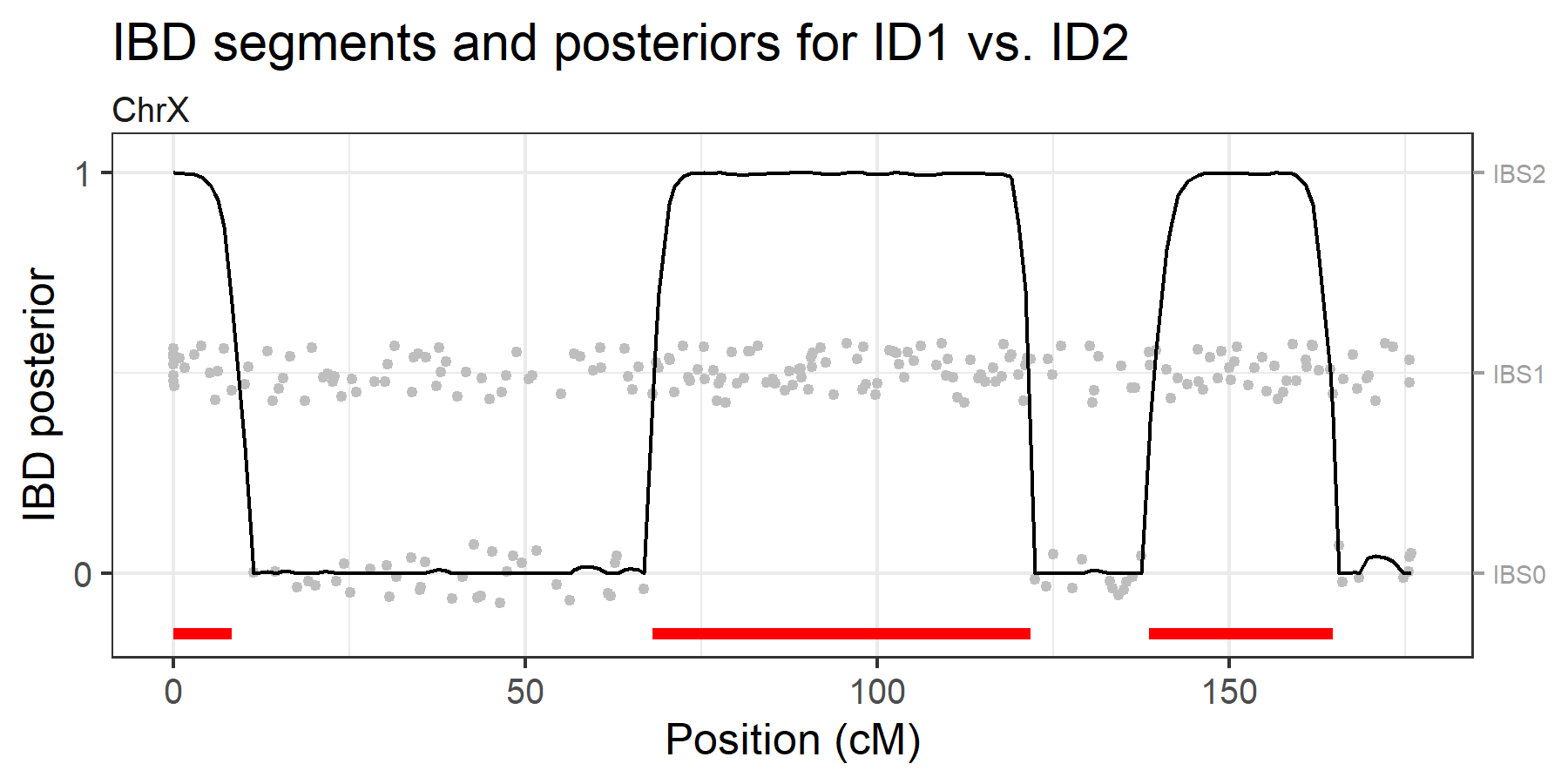
brothers typed with 246 X-chromosomal SNPs. The analysis below indicates
that they share 3 IBD segments on the X chromosome.
ibdX = findIBD(brothersX)
#> Individuals: ID1, ID2
#> Chromosome type: X (male/male)
#> Fitting HMM parameters...
#> Optimising `k1` and `a` jointly; method: L-BFGS-B
#> k1 = 0.649, a = 6.635
#> Finding IBD segments...
#> 3 segments (total length: 88.05 cM)
#> Calculating IBD posteriors...
#> Analysis complete in 0.1 secs
plotIBD(ibdX)