The goal of segregatr is to provide segregation analysis for clinical variant classification. Specifically it facilitates the calculation of full-likelihood Bayes factors (FLBs) in any medical pedigree.
If you use segregatr in a publication, please cite this paper: Ratajska et al. (2023). The use of segregation analysis in interpretation of sequence variants in SMAD3.
The paper includes applications of the package in real-life diagnostic cases.
A Shiny app shinyseg for clinical segregation analysis is now available! Check it out here: https://chrcarrizosa.shinyapps.io/shinyseg/.
The app is based on segregatr, but offers a wealth of additional features:
For details, see Carrizosa et al. (2024): shinyseg: a web application for flexible cosegregation and sensitivity analysis.
You can install segregatr from CRAN as follows:
install.packages("segregatr")Alternatively, obtain the latest development version from GitHub:
devtools::install_github("magnusdv/segregatr")library(segregatr)The family below shows four brothers, all affected with a rare dominant disease with 90% penetrance and phenocopy rate 1%. The parents have unknown affection status. All four brothers are shown to carry a candidate variant.
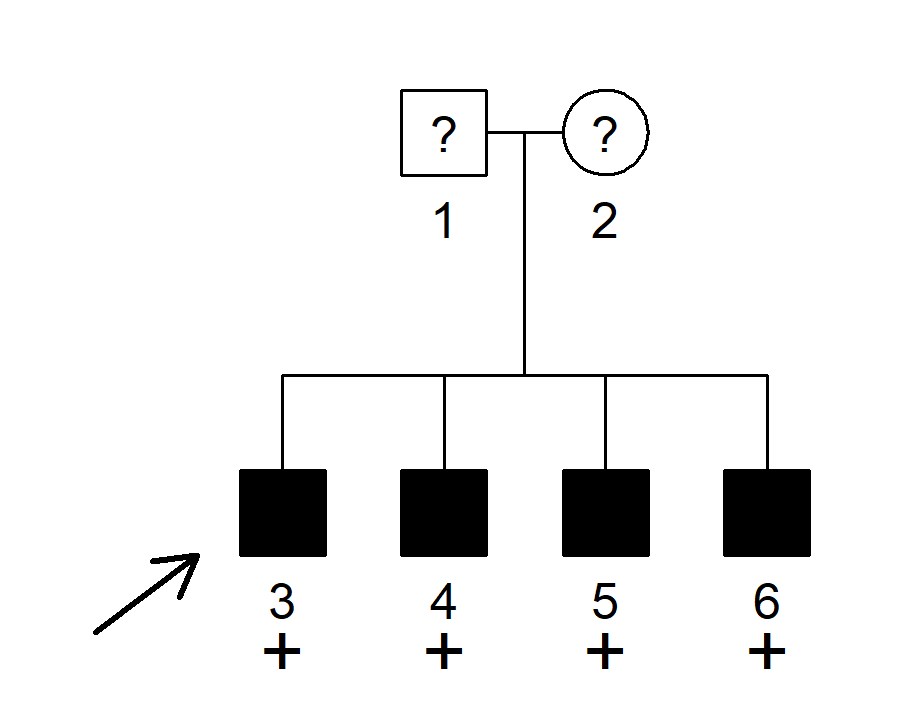
We will use segregatr to analyse the co-segregation of the variant and the disease in this pedigree. Specifically we want to compute the full-likelihood Bayes factor (FLB), quantifying the evidence that the variant is pathogenic.
To create the pedigree we use the nuclearPed() function
from the pedtools package, which is automatically
loaded together with segregatr.
x = nuclearPed(4)Then we run the FLB() function, filling in the necessary
data:
FLB(x, carriers = 3:6, affected = 3:6, unknown = 1:2,
freq = 0.0001, penetrances = c(0.01, 0.9, 0.9), proband = 3)
#> [1] 7.732161The resulting FLB score amounts to suggestive evidence for pathogenicity, according to the thresholds suggested by Jarvik and Browning (2016).